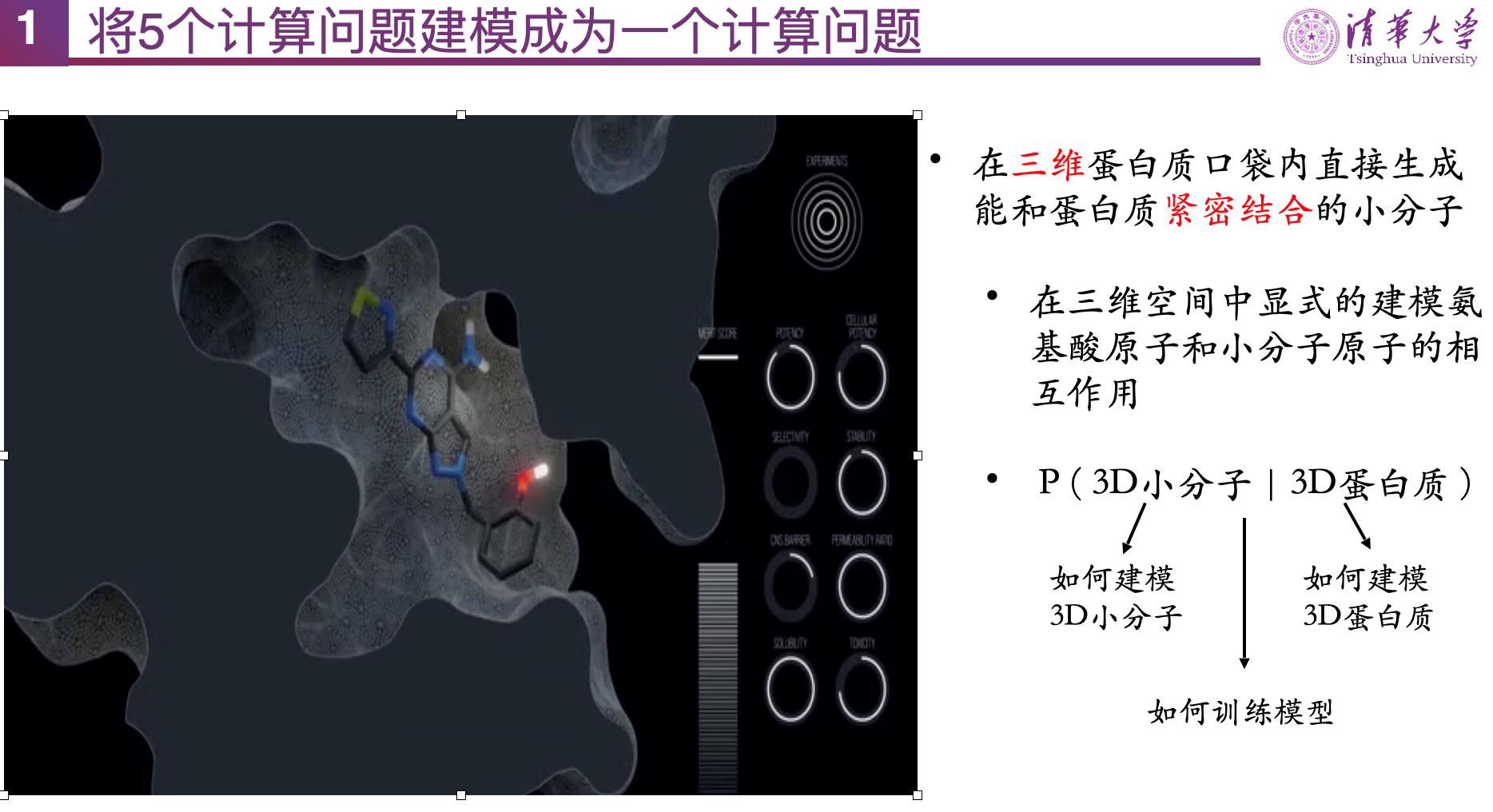
传统的分子筛选方法至少需要五步才能找到满足条件的药物候选分子,而我们把传统药物发现中的五个计算问题整合为一个计算问题,即给定靶点结构,怎样生成小分子。
10月14日下午,第5期AIR学术工作坊下半场的报告嘉宾:清华大学智能产业研究院(AIR)副教授马剑竹,为我们做了题为《基于靶点结构的小分子药物设计》的报告。

马剑竹,现任清华大学智能产业研究院副研究员/副教授,博导,海外优青项目入选者。2016年于芝加哥丰田工业研究院获得博士学位,随后在加州大学圣地亚哥分校医学院担任项目科学家。2020年开始担任美国普度大学计算机系Walther助理教授,兼任普渡大学生物化学系助理教授,2021年担任北京大学人工智能研究院副教授,北京大学公共卫生学院副教授,主要研究领域为人工智能、系统生物学、生物制药和智慧医疗。曾获得生物计算学国际顶级会议RECOMB的最佳论文奖,国际计算生物学会议ISMB的Warren DeLano奖,国际RNA和蛋白质折叠大会的最佳海报奖,论文曾被Nature Methods,Nature Machine Intelligence评为封面论文。
马剑竹教授在讲座中介绍了给定靶点设计小分子药物(Structure-based Drug Design,SBDD)的两类方案,即自回归的方法和扩散模型的方法,并介绍了它们的比较及应用。
首先,马剑竹教授从传统的虚拟筛选方法出发,说明需要经过小分子分布学习、从分布内采样、预测小分子与靶点是否结合、预测对接位姿、预测亲和力等五个计算步骤才能得到药物候选分子,效率较低,限制了找到药物候选分子的成功率。为了使 AI 更好地助力于药物设计,提升药物设计效率,马教授将多个计算问题建模为一个计算问题,即给定蛋白质口袋,生成与之紧密结合的小分子的条件概率 P(3D 小分子 | 3D 蛋白质),将问题转化为对蛋白质、小分子的分别建模以及模型训练问题,在三维空间中显式建模氨基酸原子和小分子原子的相互作用。

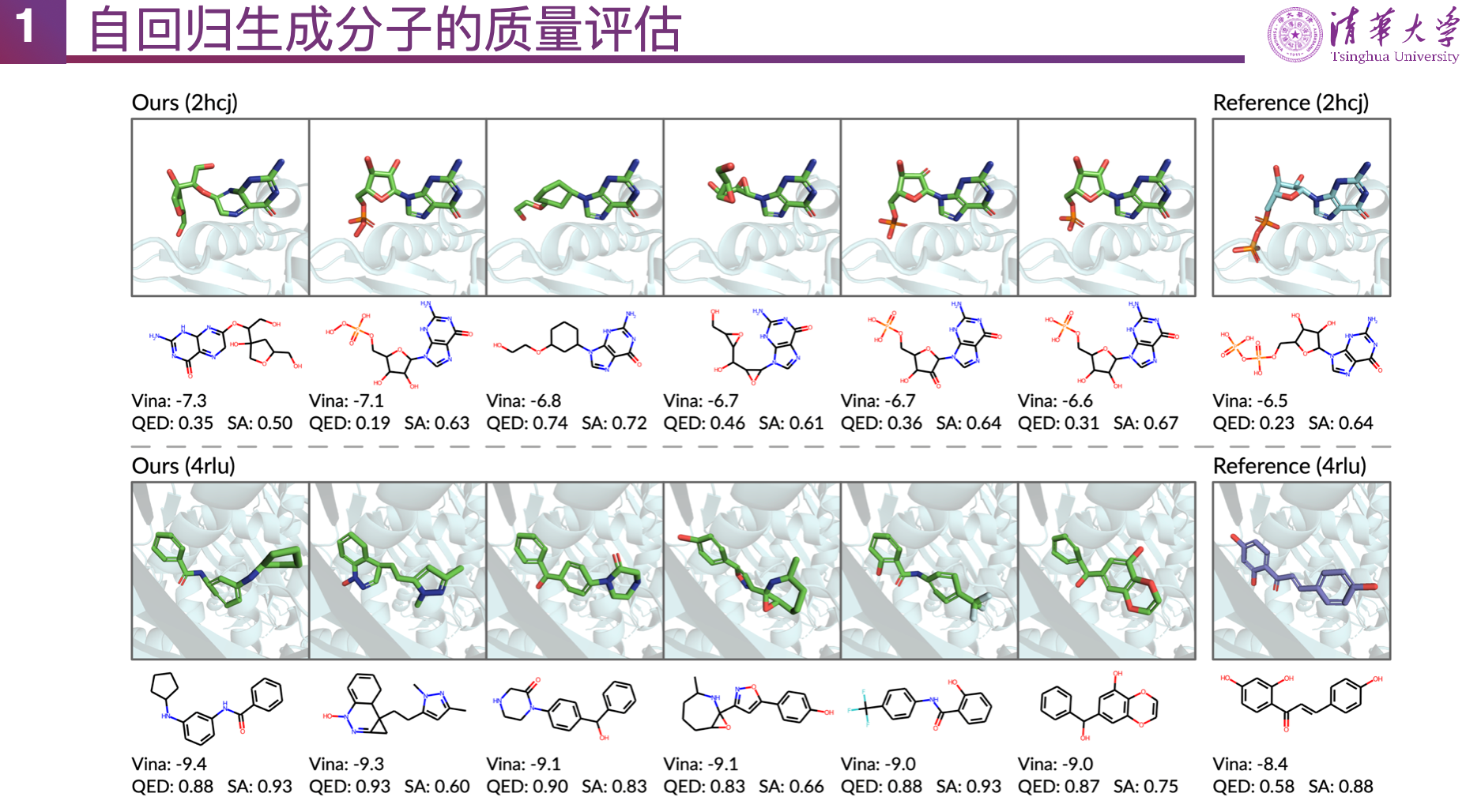
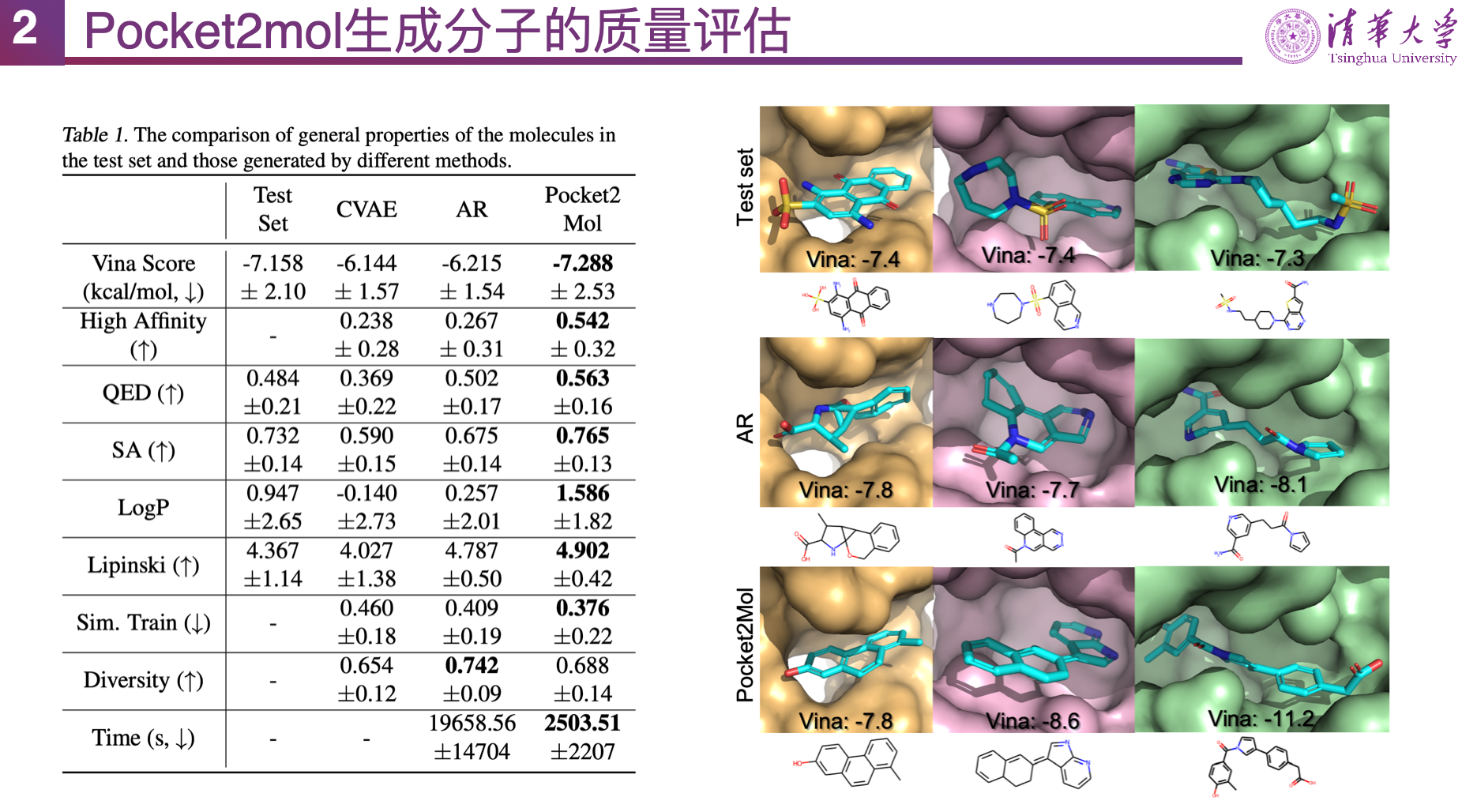
随后,马剑竹教授介绍了第一类方案,自回归生成小分子。一种方法是建模口袋内部原子密度,受到掩码语言模型(Masked Language Model)的启发,训练小分子生成模型 AR 时通过在口袋内随机遮蔽一些原子,在口袋内划分网格,预测每个格点出现某种原子的概率;生成时需要采样很多格点,再通过马尔可夫链蒙特卡洛方法(MCMC)将采样到的各格点原子概率逐个转化为原子位置和坐标,最后得到小分子。AR 模型能生成合理的结果,使用经典的分子对接软件 Vina 对结合能打分,平均在 -6.344 kcal/mol 左右,在类药性(QED)、可合成性(SA)等指标上也与训练数据分布相近。但这一模型的问题在于,打格点方式的采样效率很低。为此,马教授团队又提出了 Pocket2Mol 模型,用单独的神经网络预测中心原子(focal atom),据此进行下一个原子的位置预测,避免了网格式采样,用混合高斯提升了采样效率;此外还在逐原子的自回归生成的基础上又加入了单独的神经网络生成化学键,分子质量相比 AR 明显提升。
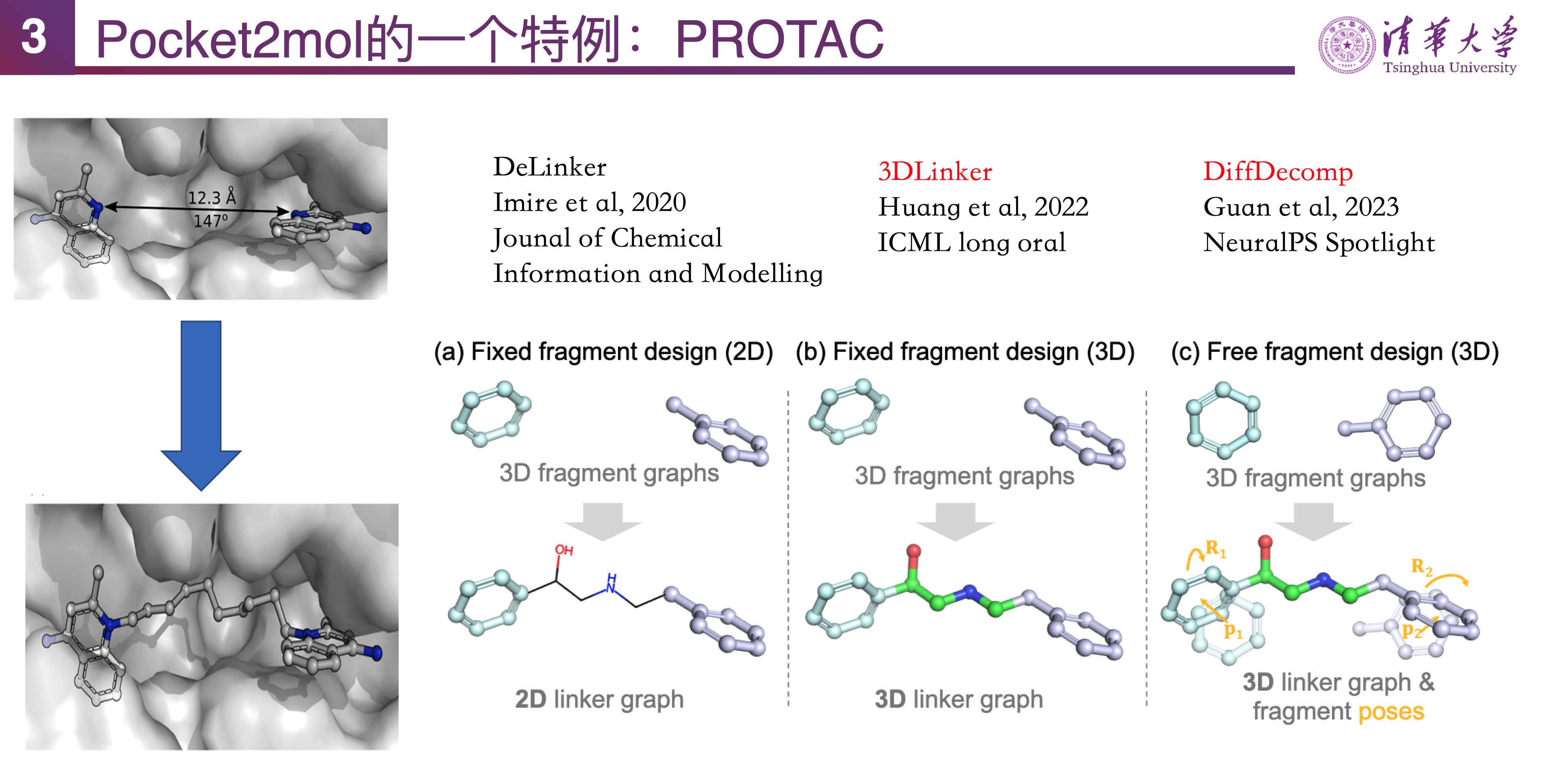
关于 Pocket2Mol 模型,马剑竹教授列举了两个应用的例子。一是以 Pocket2Mol 生成的分子为骨架,通过匹配相似的口袋结合模式进行药物筛选。剑桥大学的诺华制药团队的 Pocket Crafter 方法要求化合物库中的小分子首先在形状与静电势(electrostatics)上与生成分子匹配,其次通过一系列筛选手段,最后获得药物候选(hit)。实验结果表明,通过在 Pocket2Mol 的空间内进一步剪裁得到候选药物分子的成功率比传统筛选方法提高了十倍以上。二是蛋白分解靶向嵌合体(proteolysis-targeting chimeras,PROTAC)设计所需技术,即给定两个活性配体,设计将其连接的结构(linker),形成三联体。马教授团队提出的 3DLinker 能针对固定位置的配体片段(fragment)设计中间的连接结构,DiffDecomp 则利用一些片段作为先验,将分子设计分解为靠近口袋的结合片段、结合片段之间的连接片段,能够设计更合理、亲和力更强的分子结构。
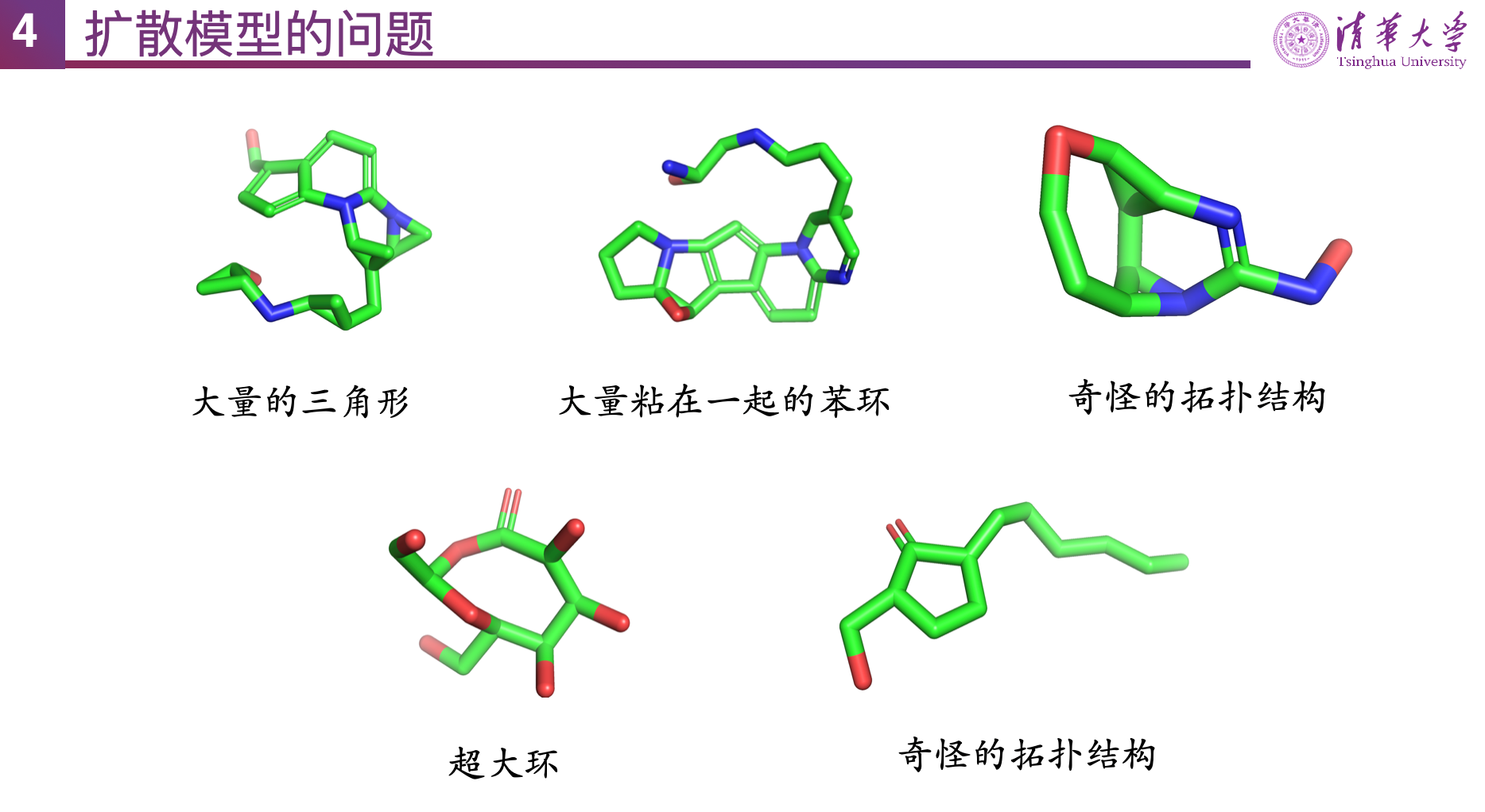
然而,Pocket2Mol 仍然存在一些问题,例如第一个原子坐标容易预测错误、生成的分子倾向于填满口袋内整个空间、难以预测何时停止等,这是由自回归的生成方式造成的。因此,马剑竹教授团队进一步探索了第二类方案,基于扩散模型的非自回归小分子生成模型。TargetDiff 所有原子一起生成,每个时刻都有全局信息,这样生成的分子键长和键角更符合训练数据。得益于扩散模型的高效训练,模型除了生成分子之外,对分子的似然还可以被用于结合能预测,相关系数可达 0.4-0.5。但是,由于 TargetDiff 仅在原子类型和坐标上进行扩散,通过 OpenBabel 后处理添加化学键,生成的分子构象仍然不够合理,有的分子存在三元环、并环、超大环以及其他可能扭曲的结构问题。
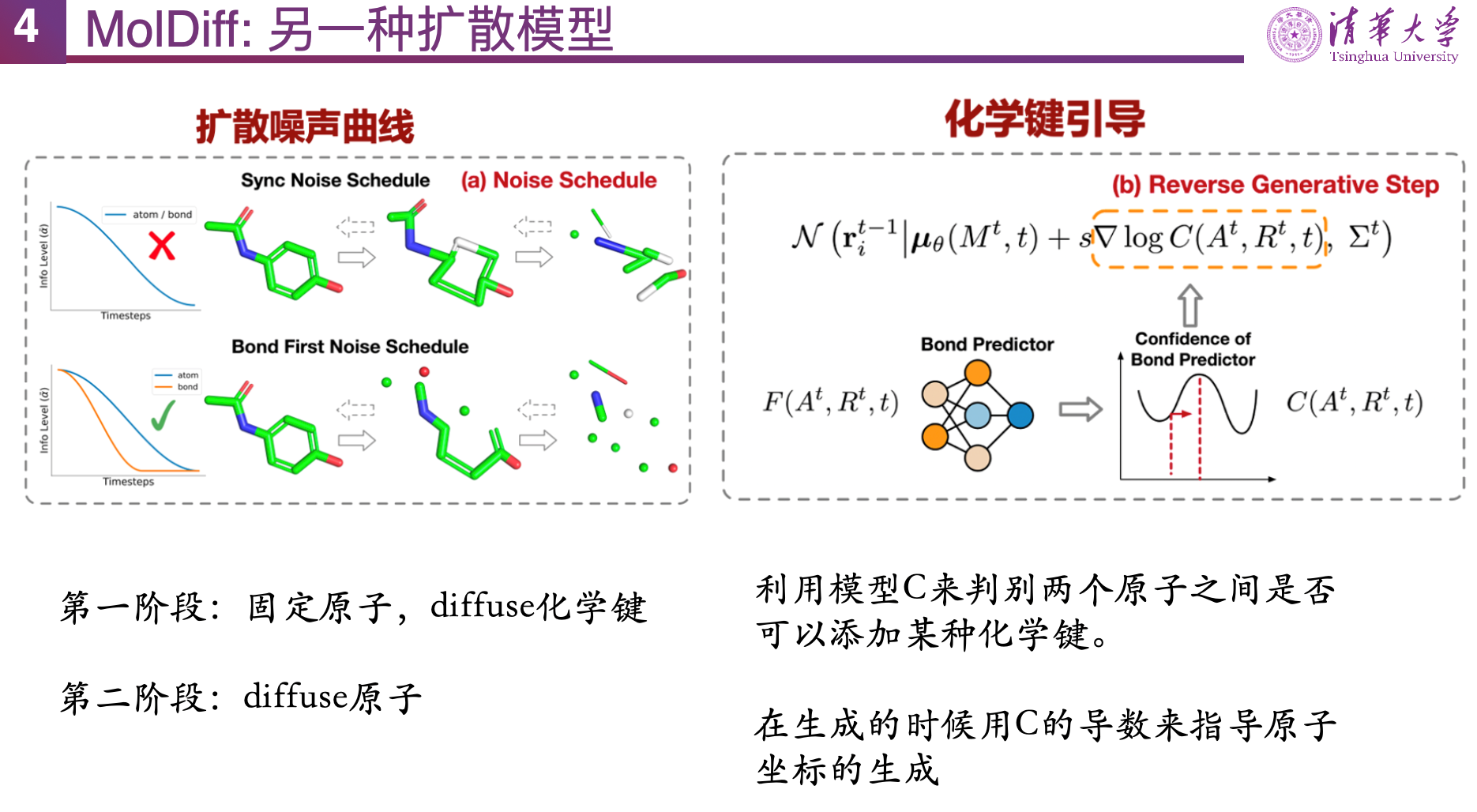
扩散模型如何更好地适配分子数据的模态?马剑竹教授进一步考虑到分子拓扑结构,认为不能和图片一样纯粹对原子坐标和类型进行加噪,而应该和加噪后的图片仍像一张图片一样,考虑如何加噪使得分子仍然像一个分子。马教授团队的 MolDiff 将扩散过程设计为两个阶段,即化学键生成和原子生成,在加噪时先破坏化学键,再给原子加噪,去噪时先生成原子,再加入化学键的生成及引导,解决了 TargetDiff 中存在的一些问题。MolDiff 生成分子的连通性、生成成功率均提升至 0.99 以上,成功率提升近三倍,大幅提高了生成能力。在生成质量上,从类药性、拓扑结构和环的特征等多个质量评估角度也都取得明显提升。
最后,马剑竹教授对上述方案进行了比较,目前的结论是 Pocket2Mol 仍然是十分有力的基于靶点结构的分子生成模型。马教授希望,在未来探索中能进一步结合各个模型的优势,助力药物设计领域发展。